Molecular Factories#
The molecular_factories package provides a collection of tools for the automatic generation of molecular structures.
Specifically implemented are:
The Assembler class is a class that can be to assemble molecules from a library of fragments.
It requires a list of Molecules that serve as the fragments to be assembled. The class will then generate random molecules by randomly selecting fragments from the library and attaching them to each other.
Usage#
Create a list of fragments to be used for assembly
Create an instance of the Assembler class with the list of fragments
Use the sample method to generate random molecules or the make method to create a specific fragment from an instruction matrix
Example
Let’s make a little toy example
import buildamol as bam
from buildamol.extensions.molecular_factories import Assembler
import matplotlib.pyplot as plt
# get some molecules to serve as fragments
fragments = [
bam.Molecule.from_smiles("C1=CC=CC=C1", id="A").autolabel(),
bam.Molecule.from_smiles("CC=O", id="B").autolabel(),
bam.Molecule.from_smiles("COC=C", id="C").autolabel(),
bam.Molecule.from_smiles("C1=CCC=C1", id="D").autolabel(),
bam.Molecule.from_smiles("C(C)N", id="E").autolabel(),
]
# make the assembler
assembler = Assembler(fragments)
# generate some molecules from 3 fragments each
# let's make 9 molecules
molecules = assembler.sample(n_fragments=3, n=9)
fig, axs = plt.subplots(3, 3, figsize=(12, 12))
for mol, ax in zip(molecules, axs.flat):
ax.imshow(
mol.draw2d().draw(),
)
ax.axis("off")
plt.show()
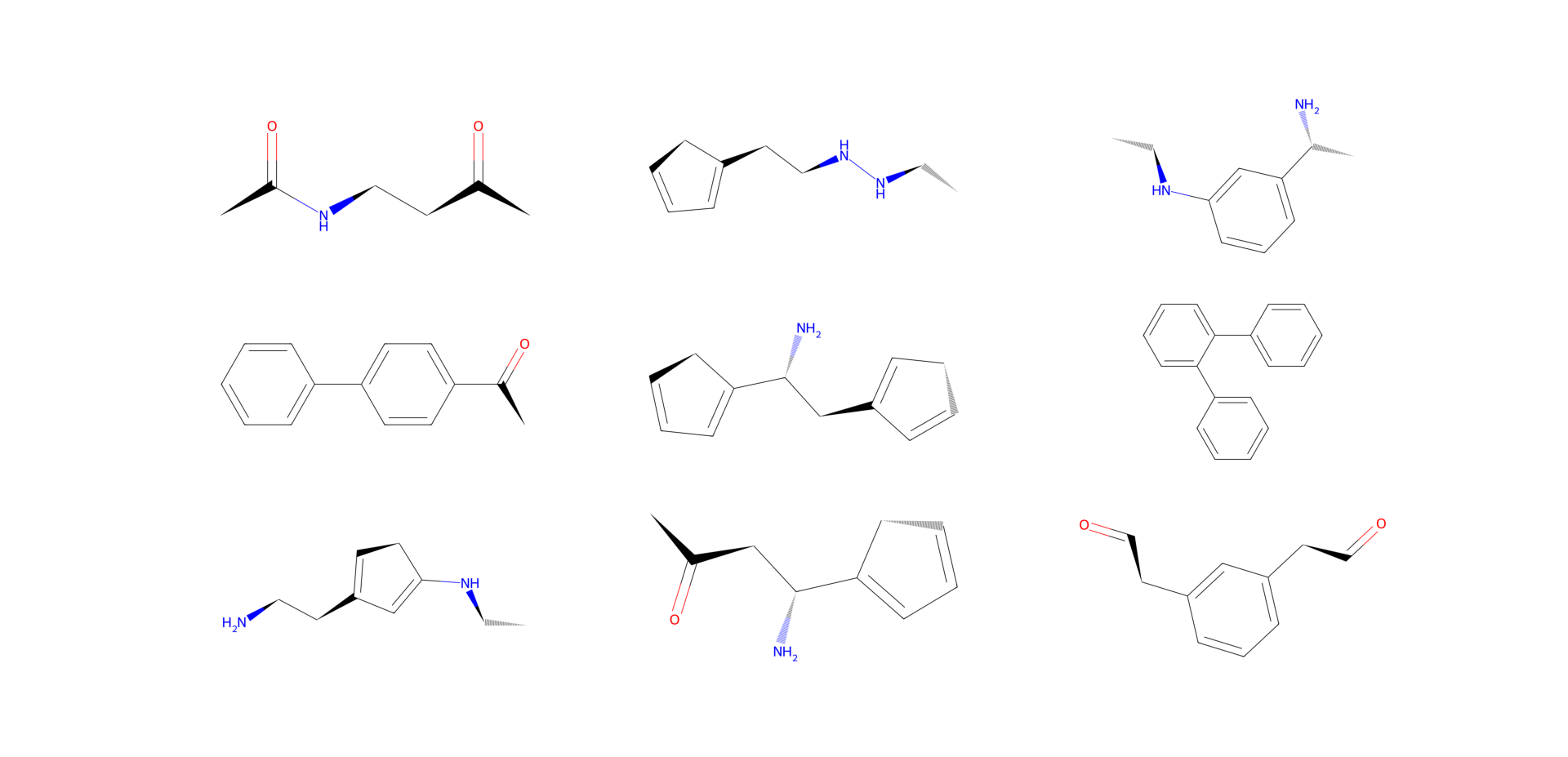
Making Molecules from Arrays#
We can also use the make method to create a specific molecule from an instruction matrix This matrix is a 2D numpy array where each row corresponds to an instruction for attaching the next fragment onto the molecule. The columns are as follows:
[
[incoming_fragment_global_index, incoming_atom_index, target_fragment_atom],
[incoming_fragment_global_index, incoming_atom_index, target_fragment_atom]
...
]
The incoming_fragment_global_index is the index of the fragment in the fragment library (i.e. in the list). The incoming_atom_index is the index of the atom in the incoming fragment that will be attached to the target fragment (i.e. the attachment point). The target_fragment_atom is the index of the atom in the target fragment that will be attached to the incoming fragment.
Let’s make a molecule from an instruction matrix. Let’s take the fourth fragment molecule as a start. Then attach the second fragment molecule to it, by attaching the its second atom to the first atom of already present molecule. Then attach again the fourth fragment onto the molecule by attaching its first atom to the first atom of the second fragment in the molecule.
matrix = np.array([
[3, 0, 0],
[1, 1, 0],
[3, 0, 0],
])
mol = assembler.make(matrix)
mol.draw2d().show()
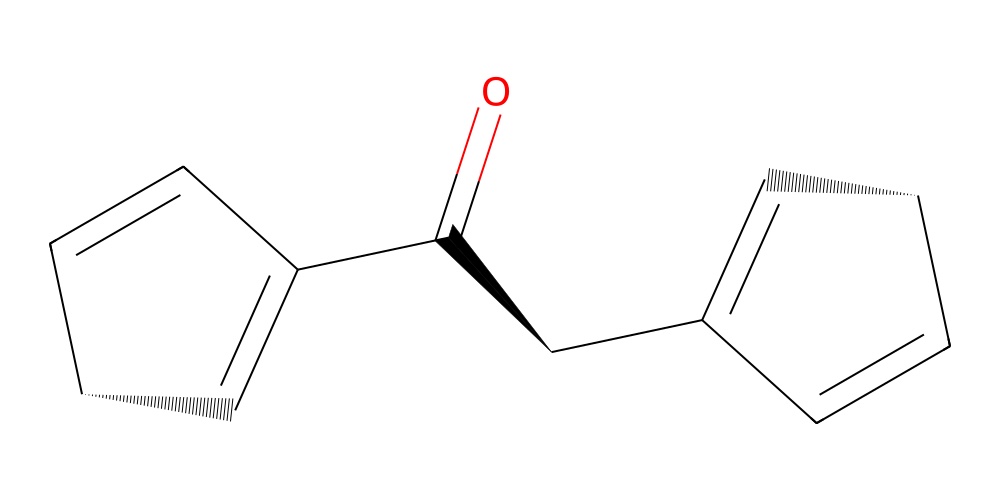
If including this into an automatic pipeline or an optimization loop it is recommended to wrap the whole thing into a try-except block to catch any errors that might occur due to invalid matrices. The clue is that the atoms used for attachment should not be used more than once in the matrix. If they are used more than once, the molecule will not be able to be assembled leading to an error.
- class buildamol.extensions.molecular_factories.assembler.Assembler(fragments: list)[source]#
Bases:
objectThe Assembler class is a class that can be to assemble molecules from a library of fragments. Each molecule is a linear chain of fragments that are attached to each other.
- Parameters:
fragments (list) – A list of Molecules that serve as the fragments to be assembled.
- make(matrix: ndarray) Molecule[source]#
Assemble a molecule based on an instruction matrix
- Parameters:
matrix (np.ndarray) – The matrix encoding for the molecule
- Returns:
The assembled molecule
- Return type:
- random(n_fragments: int) ndarray[source]#
Make a random matrix encoding for a molecule assembled from fragments.
- Parameters:
n_fragments (int) – The number of fragments to use for the molecule
- Returns:
A matrix encoding for the molecule
- Return type:
np.ndarray
- sample(n_fragments: int, n: int = 1)[source]#
Generate n random molecules from the fragment library
- Parameters:
n_fragments (int) – The number of fragments to use for each molecule
n (int) – The number of molecules to generate
- Yields:
Molecule – A molecule assembled from the fragments
- specify_attachment_points(fragment_or_index, points: list)[source]#
Specify the attachment points for a fragment
- Parameters:
fragment_or_index (int or Molecule) – The fragment for which to specify the attachment points
points (list) – The attachment points to specify. These must be the indices of the atoms in the fragment as they appear in fragment.get_atoms() (NOT the serial_number!).
The Derivator class can be used to create stochastic and/or combinatorical derivatives of a molecule.
Usage#
Create a Derivator object with a molecule as an argument.
Use the element_changable, functional_group_addable, bond_order_changable, and modifiable methods to specify the possible changes that can be made to the molecule.
Use the sample, all, or make method to generate one or more derivative molecule(s).
Example
Let’s say we have a benzene ring which we want to derivatize. Let’s say further that we are interested in changing the elements of two atoms and adding one functional group to an atom. We can do this as follows:
Create the benzene molecule and derivator object
import buildamol as bam
bam.load_small_molecules()
benzene = bam.molecule("benzene")
# Create the derivator object
D = bam.Derivator(benzene)
Specify the possible changes that can be made to the molecule
Let’s say that C1 can be either a carbon, a nitrogen, or an oxygen. And C5 can be either a carbon or a nitrogen. Also, we are interested in possibly adding a functional group to C3. Namely, we want to check with a hydroxyl group and a phosphate group.
# set the possible elements for C1 and C5
D.element_changable(
"C1",
("C", "N", "O"),
)
D.element_changable(
"C5",
("C", "N"),
)
# set the possible functional groups for C3
# (the groups are functions that modify the molecule in place)
D.functional_group_addable(
"C3",
(D.nothing, bam.hydroxylate, bam.phosphorylate),
)
Generate derivatives
Since we only have a small number of changes, we can generate all possible derivatives using the all method.
# generate all possible derivatives
derivatives = list(D.all())
Visualize the derivatives
Now we can visualize the derivatives using the draw2d method.
import matplotlib.pyplot as plt
# compute the layout for the plots (looks nicer like this)
rows = np.sqrt(len(derivatives))
cols = np.ceil(len(derivatives) / rows)
rows = np.round(rows)
fig, axs = plt.subplots(int(rows), int(cols), figsize=(12, 12))
for i, molecule in enumerate(derivatives):
molecule.squash()
try:
img = molecule.draw2d().draw()
axs.flat[i].imshow(img)
except Exception as e:
print(e)
for ax in axs.flat:
ax.axis("off")
plt.show()
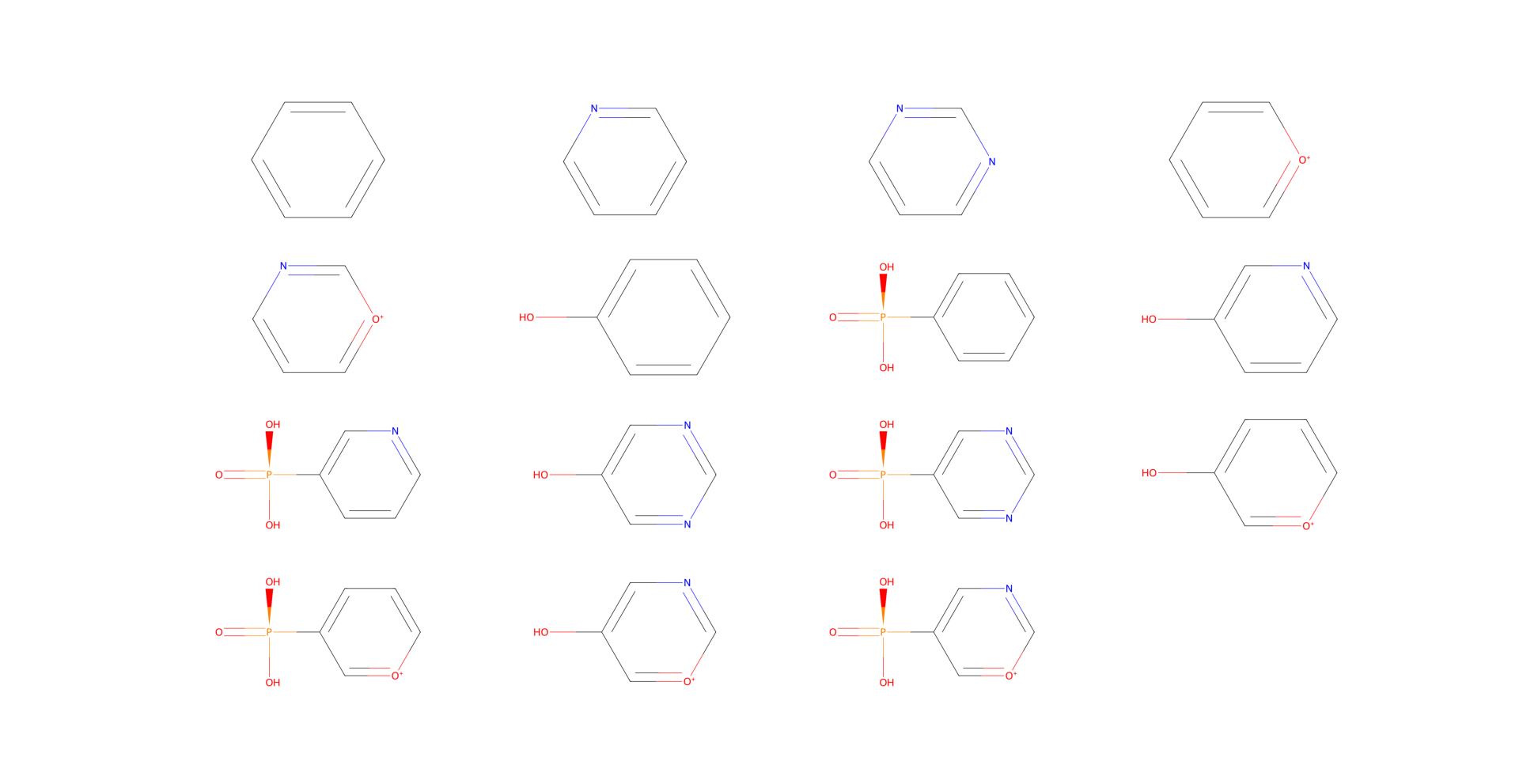
- class buildamol.extensions.molecular_factories.derivator.Derivator(molecule: Molecule)[source]#
Bases:
objectA class to generate derivatives of a molecule.
- Parameters:
molecule (Molecule) – The molecule to generate derivatives of
- property N: int#
The number of possible derivatives
- all() Generator[Molecule][source]#
Generate all possible derivatives
- Yields:
Molecule – The generated derivative molecule
- bond_order_changable(bond, bond_orders: tuple, probabilities: tuple = None)[source]#
Specify a bond to be derivable to a set of possible bond orders
- Parameters:
bond (Bond) – The bond
bond_orders (set) – The set of possible bond orders
probabilities (tuple) – The probabilities of each bond order being used
- element_changable(atom, elements: tuple, probabilities: tuple = None)[source]#
Specify an atom to be derivable to a set of possible elements
- Parameters:
atom (Atom) – The atom
elements (set) – The set of possible elements
probabilities (tuple) – The probabilities of each element being used
- functional_group_addable(atom, group_modifiers: tuple, probabilities: tuple = None)[source]#
Specify an atom onto which a functional group can be added
- Parameters:
atom (Atom) – The atom
group_modifiers (tuple) – The set of possible functional groups that can be added onto the atom. Each entry must be a function that will modify the molecule in place, taking only the molecule and target atom as arguments!
probabilities (tuple) – The probabilities of each functional group being added
- global_modifiers(modifiers: tuple, probabilities: tuple = None)[source]#
Specify a set of possible modifications to the molecule as a whole.
- Parameters:
modifiers (tuple) – The set of possible modifications that can be applied to the molecule. Each entry must be a function that will modify the molecule in place, taking only the molecule as an argument!
probabilities (tuple) – The probabilities of each modification being applied
- make(elements: tuple, bonds: tuple, groups: tuple, modifiers: tuple) Molecule[source]#
Make a specific derivative molecule. Provide a tuple or other ordered iterable of the same length as the number of atoms/bonds specified during setup for each editable category. Each element must be an integer denoting the index of the desired derivative application.
- Parameters:
elements (tuple) – The elements to apply.
bonds (tuple) – The bond orders to apply.
groups (tuple) – The group modifiers to apply.
modifiers (tuple) – The molecule modifiers to apply.
- Returns:
The generated derivative molecule
- Return type:
- sample(n: int = 1, use_probabilities: bool = True, apply_all_modifiers: bool = False) Generator[Molecule][source]#
Create a derivative molecule
- Parameters:
n (int) – The number of derivatives to generate
use_probabilities (bool) – Whether to use the probabilities when generating derivatives
apply_all_modifiers (bool) – If True all modifiers are applied to the sampled molecule(s). Otherwise one modifier is sampled and applied to each molecule.
- Yields:
Molecule – The generated derivative molecule