Basic Usage#
Most of the functionality is provided through a simple toplevel functional API, with a much more extended method-based API available for both simple and more complex operations. To facilitate user-friendliness most of the biobuild functionality has been integrated into methods that are attached to the Molecule class, which is the main class of biobuild. The Molecule class is a wrapper around the Bio.PDB.Structure.Structure class which handles all atomic data.
Note
The Molecule class is not a subclass of Bio.PDB.Structure.Structure - it is a wrapper around it. This means that it does not inherit any methods of the Structure class directly. However, the Molecule class has a structure attribute which is the Structure object that it wraps. Also, the Molecule class is equipped with many equivalent methods to the Structure class that can be used to access the structure in more convenient ways.
The Molecule class is the main class of biobuild and is used to create, modify, and visualize molecular structures. The Molecule class can:
read structures from PDB and mmCIF files
write structures to PDB and mmCIF files
convert structural data between formats
add and remove atoms, residues, and chains
rotate parts of the structure around bonds
connect multiple molecules together
visualize structures in 3D
provide structural data such as bond angles
import biobuild as bb
# make a molecule from a PDB file
mol = bb.molecule("my_structure.pdb")
# rotate part of the molecule
# around the bond between atoms C4 and C5 by 45°
# rotating only atoms after C5 (i.e. descendants)
mol.rotate_around_bond("C4", "C5", 45, descendants_only=True)
# write the molecule to a mmCIF file
mol.to_cif("my_structure.cif")
Did you know?
The toplevel molecule function can be fed with a variety of inputs and will automatically try to discern the correct processing
in order to produce a Molecule object. Namely, the following inputs are all valid:
a PDB or CIF filename
a PDB id
a molecule name (e.g. “alpha-d-glucose”)
a SMILES string
an InChI string or key
a Structure object from Bio.PDB
Functional API#
The toplevel functional API is the most straightforward one, and is for many operations the easiest one.
Functions such as read_pdb or polymerize are intuitive and easy to use. However, the functional API is
not as extensive as the method-based API, since it simply calls the method-based API under the hood. Many other
operations are therefore not available in the functional API - for instance, removing atoms from a molecule is only available
in the method-based API.
import biobuild as bb
# load sugar data repository
bb.load_sugars()
# make a molecule from a
# defined chemical compound
glc = bb.molecule("alpha-d-glucose")
# polymerize the glucose molecule
# to make cellulose
cellulose = bb.polymerize(glc, 100, linkage="14bb")
# write the cellulose to a PDB file
bb.write_pdb(cellulose, "cellulose.pdb")
Method-based API#
The method-based API allows for more flexibility. At the heart of biobuild is the Molecule class, which
has a great number of methods to modify the structure or add/remove parts of it. Methods such as get_atom,
add_bond, or remove_residue are basic operations to modify a structure in-place, while methods such as attach
allow to expand the structure by adding new parts to it.
import biobuild as bb
bb.load_sugars()
# make a molecule from a
# defined chemical compound
glc = bb.Molecule.from_compound("alpha-d-glucose")
# polymerize the glucose molecule
# to make cellulose
cellulose = glc.repeat(100, linkage="14bb")
# write the cellulose to a PDB file
cellulose.to_pdb("cellulose.pdb")
Operator-based API#
The operator-based API is a short-hand proxy to the method-based API (just as the functional API is a proxy). It is essentially restricted to operations that regard connecting two molecules together. However, it is the most condensed way to write biobuild code - sometimes at the expense of readability. Available operators are:
+ for connecting two molecules together
* for polymerizing a molecule
% for specifying the linkage between two molecules
@ for specifying the residue at which to create a connection between two molecules
^ for specifying the atom to use for a connection (more detailed than @)
Note
In-place versions of the operators are also available, e.g. += for connecting two molecules in-place, or *= for in-place polymerization.
import biobuild as bb
bb.load_sugars()
glc = bb.Molecule.from_compound("alpha-d-glucose")
# polymerize the glucose molecule into cellulose
cellulose = glc % "14bb" * 100
# write the cellulose to a PDB file
cellulose.to_pdb("cellulose.pdb")
Built-in-resources#
biobuild comes with a number of built-in data resources. Namely, biobuild integrates the PDBE component library for
components up to 40 atoms in size by default - naturally, the full library can be loaded if desired. This enables molecule creation through the from_compounds method that can be queried using PDB id, chemical name, SMILES, InChI and InChIKey.
Furthermore, biobuild integrates parts of the CHARMM force field for
references of molecular connections. You may have noticed that in the above examples, the 1->4 beta glycosyidic linkage was used a lot, but only referred to as "14bb".
This is because the CHARMM force field has the geometric data stored under this identifier.
Finally, biobuild integrates pubchempy for the direct retrieval of molecules from PubChem (requires internet connection).
Toplevel functions exist to access these resources, e.g. biobuild.available_linkages() to get a list of pre-defined linkages,
or biobuild.has_compound("alpha-mannose") to check if a particular compound is available in the loaded PDBE component library. Also,
in order to make biobuild more useful to the respective user, it is possible to add custom data to the standard resources and set new default settings
using functions such as set_default_topology or add_linkage.
Example#
Building a glycan#
biobuild was originally conceptualized with the aim of creating glycan structures - so, please, forgive if the example below produces a glycan. The following example demonstrates how we can create a larger structure from single monosaccharides using biobuild (using all three syntaxes intermixed):
import biobuild as bb
bb.load_sugars()
# get the monosaccharides
# (using their PDBE identifiers)
nag = bb.molecule("NAG") # N-acetylglucosamine, a.k.a. GlcNAc
bma = bb.molecule("BMA") # beta-mannose
man = bb.molecule("MAN") # alpha-mannose
# start by connecting two NAGs together
# 'beta 1->4' glycosydic linkage is pre-defined
# in the CHARMM force field and can be used by its name '14bb' directly
glycan = nag % "14bb" + nag
# add a beta-mannose to the last NAG
glycan += bma
# add an alpha-mannose to the beta-mannose
# using an 'alpha 1->3' linkage ('13ab' in CHARMM)
glycan.attach(man, "13ab")
# add another alpha-mannose
# at the second-to-last residue (BMA)
glycan.attach(man, "16ab", at_residue=-2)
# add one final alpha-mannose
glycan = bb.connect(glycan, man, "16ab")
# now visualise the structure
glycan.show()
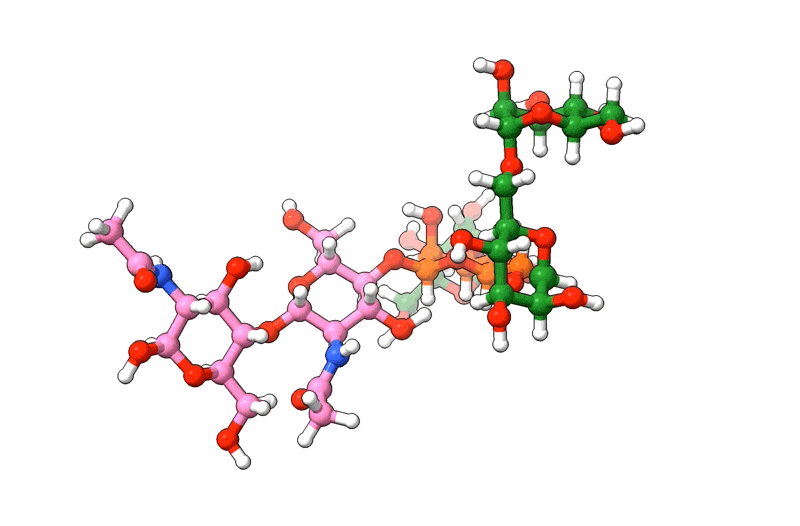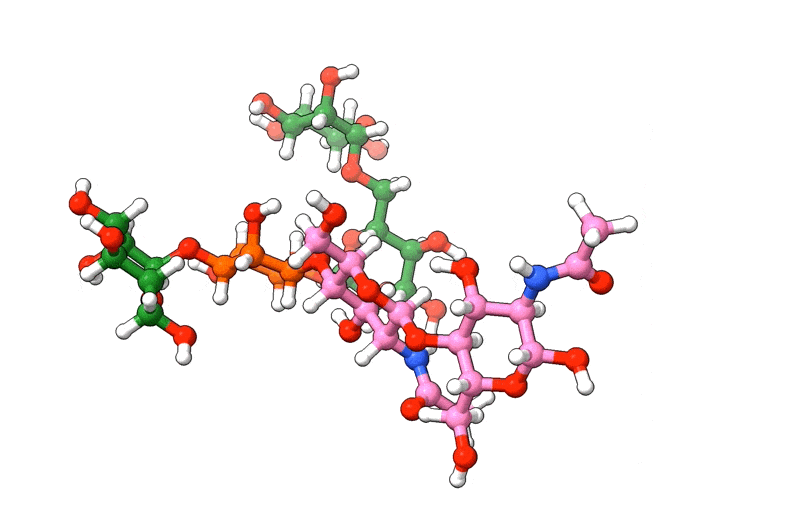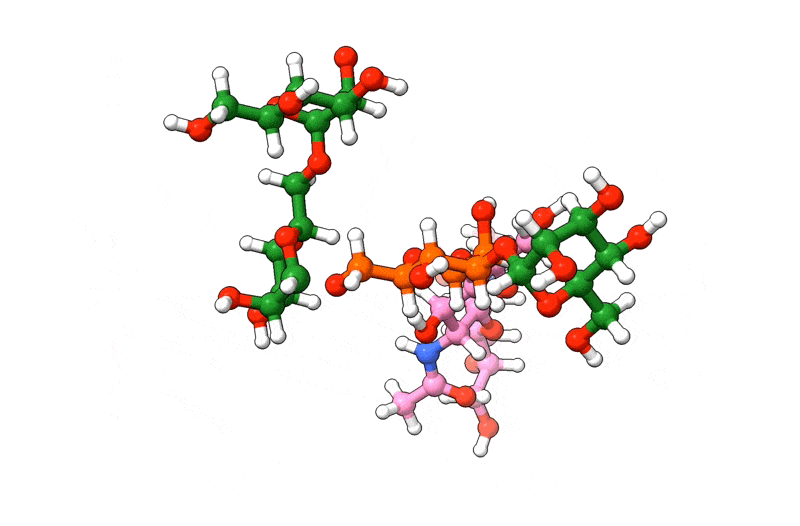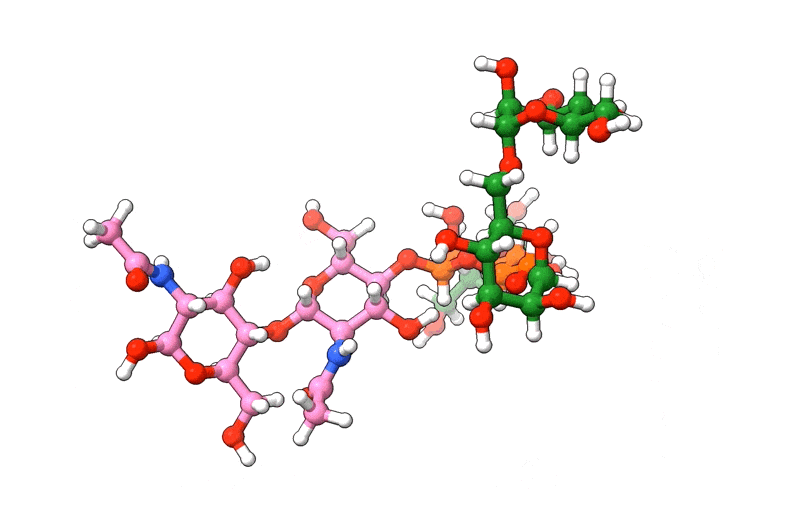
In the above visualization, NAG residues are colored in pink, BMA in orange, and MAN in green. Hetero-atoms are colored according to IUPAC conventions.
The above example demonstrates how we can use biobuild to create a glycan structure from scratch. The example also demonstrates how we can use the three different syntaxes to achieve this. Using the toplevel function connect, using the method attach, or by simple “molecular arithmetics” through the + operator.