Making a Rotaxane (hard)#
In this tutorial we will cover:
how we can use BuildAMol when we want to setup an entire multi-molecule system
how we can use BuildAMol to fit a molecule to a particular position in space
So, presumably you have checked out the “easy” rotaxane tutorial and are now wondering what the “hard” version would look like? Well, the primary difference is that we are here not relying on the RotaxaneBuilder to align the ring around the scaffold but instead we are using a Translatron optimization environment together with a self-defined constraint function to perform positional fitting ourselves.
Sounds like a headache? Well, yes, a little, but in exchange, you will learn how to create (potentially) any kind of multi-molecule system that contains multiple molecules in some spacial arrangement. The same principles that we use here to make a rotaxane can be used to make complexes or cages or frameworks. It only depends on your creativity and the constraint function you design… and a bit of luck since we are using optimizations here, so every round has the potential to go bogus…
Anyway, with all that said, let’s dive into our rotaxane makery… Again, like last time we are building this compound below from a paper by Tian et al. (2020)
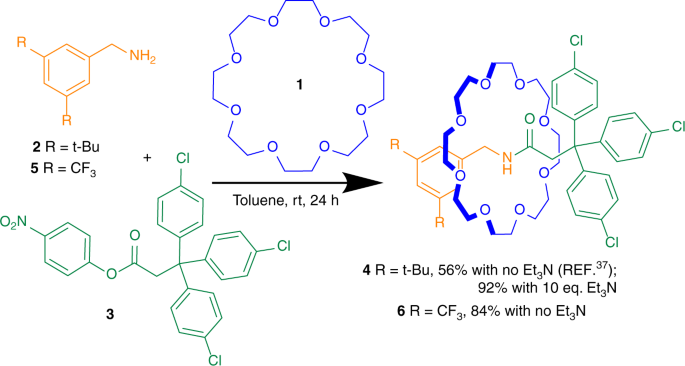
As we can see, the final compound on the right consists of two individual molecules. One circular structure (blue) and the scaffold (green and orange).
We will first build the components separately and then assemble a system for PDB export from these.
[1]:
import plotly
plotly.offline.init_notebook_mode()
[2]:
import buildamol as bam
Loading the ring and scaffold#
In the previous tutorial we made the ring and scaffold already so we can just re-use the components here.
[3]:
# load the molecules
ring = bam.molecule("./files/rotaxane_ring.json")
scaffold = bam.molecule("./files/rotaxane_scaffold.json")
v = scaffold.draw(show_atoms=False) + ring.draw(show_atoms=False, line_color="red")
v.show()
Now that we have both the ring and scaffold ready, let’s prepare a system with the molecules.
To do that we need to move the ring so that it is position around the “neck” part of the scaffold. Remember, in the last tutorial we named the “neck” residue “NCK”? That’s so we could more easily identify where to place the ring’s center.
Using the Translatron#
Fitting molecules into a specific location is not exactly BuildAMol’s core business but there is a special type of optimizer that can do so, named the Translatron. The Translatron accepts a constraint_function which it tries to minimize by translating and rotating the molecule in space.
Therefore as long as we define a proper constraint function we should be able to fit our molecule automatically onto a good position. At least in theory… Let’s see it in practice:
[60]:
import numpy as np
from scipy.spatial.distance import cdist
# get the center of the neck
# (let's just use the nitrongen atom at the center of the NCK residue for this purpose)
center = scaffold.get_atom("N4").coord
# and move the ring so that the center is at the center of the neck
ring.move_to(center)
# ---------------------------------------------------
# now define the constraint function that the
# center of the ring should be at the center of the neck
# ---------------------------------------------------
# first get the coordinates of both ends of the scaffold (the parts with the benzene rings attached to them)
# (if you don't remember which residues are where in the scaffold, be sure to revisit the last tutorial and look at the scaffold molecule)
coords1 = np.concatenate([i.get_coords() for i in scaffold.get_residues(1, 2, 3)])
coords2 = np.concatenate([i.get_coords() for i in scaffold.get_residues(-1, -2, -3)])
dist_12 = cdist(coords1, coords2).mean()
# then define the constraint function
def constraint(env, coords):
# the ring should be placed so that the distances to both ends
# are about equal
dists1 = cdist(coords, coords1)
dists2 = cdist(coords, coords2)
d1 = dists1.mean(axis=1)
d2 = dists1.mean(axis=1)
diff = (d1.mean() - dist_12 / 2) ** 2 + (d2.mean() - dist_12 / 2) ** 2
# # we want the ring to be close to the center of the neck
_d1 = d1.min()
_d2 = d2.min()
diff2 = 0 #(_d1 - _d2) ** 2
# also we don't want to have too many clashing atoms
_c = ((dists1 < 3).sum() + (dists2 < 3).sum()) ** 8
_d = _d1 + _d2
env._d = _d
# return the constraint value
return diff + diff2 - _d1 - _d2 + _c
# let's also define a function that tells us when we are done
def done(env, state):
return env._d > 5.5
Okey, with all of this set up, we can actually place the ring around the scaffold molecule using a Translatron. Here’s how we do it:
[67]:
# create a Translatron for optimization
# (this is very similar to setting up a Rotatron)
# (the bounds=(0, 0) tells the Translatron that we don't want to fit a translation as well, only the rotation)
graph = ring.get_atom_graph()
translatron = bam.optimizers.Translatron(graph, constraint_func=constraint, finish_func=done, bounds=(0, 0))
# and now we can optimize the ring placement
# (unfortunately we cannot use the toplevel "optimize" function here because
# that one is tailored to Rotatrons and cannot work with the Translatron)
solution, _ = bam.optimizers.scipy_optimize(translatron)
# now apply the solution by setting the coordinates of the ring atoms
# (the first three values are the translation, which we didn't do here,
# the last three are the rotation angles around the x, y, and z axis)
ring_positioned = ring.copy()
ring_positioned.rotate(solution[3], "x", angle_is_degrees=False)
ring_positioned.rotate(solution[4], "y", angle_is_degrees=False)
ring_positioned.rotate(solution[5], "z", angle_is_degrees=False)
ring_positioned.move_to(center)
/Users/noahhk/anaconda3/envs/glyco2/lib/python3.11/site-packages/gym/spaces/box.py:73: UserWarning:
WARN: Box bound precision lowered by casting to float32
[67]:
Molecule(ALK)
Cool, let’s inspect how the placement actually turned out:
[68]:
v = scaffold.draw()
v += ring_positioned.draw(show_atoms=False, line_color="blue")
v.show()
/Users/noahhk/anaconda3/envs/glyco2/lib/python3.11/site-packages/plotly/express/_core.py:1985: FutureWarning:
When grouping with a length-1 list-like, you will need to pass a length-1 tuple to get_group in a future version of pandas. Pass `(name,)` instead of `name` to silence this warning.
Now that looks quite descent! Let’s take it! (actually, it took a bunch of re-runs to get to this one…)
Now the last thing that’s left to do is to merge the two molecules into one object. That’s exactly the same as before:
[69]:
scaffold.merge(ring_positioned)
[69]:
Molecule(BNZ)
And with this we are done! We can export our system to a file and now go on doing whatever we like with it.
[70]:
scaffold.to_molfile("./files/rotaxane_hard.mol")
And with that we have reached the end of this tutorial. Hopefully you now know hot wo create multi-molecule systems in BuildAMol by making individual molecules, merging them together and also fitting placements to them using the Translatron optimization environment. Thanks a lot and good luck with your research!