Functional Groups#
In this tutorial we will cover:
how we can use functional groups in BuildAMol to connect molecules
NOTE:
The functional groups that are discussed here have been succeeded by the new
Reactivityclasses that offer a greater flexibility and easy-of-use. If you are interested in learning more about BuildAMol+Chemistry and you are not already invested in using the functional groups it is recommended to check out the tutorial on the Reactivities instead! You can always come back to this tutorial if the Reactivities are not for you :-)
BuildAMol offers great flexibility when connecting molecules. It is designed create any kind of single-bond between any two atoms and is not restrained to work in the realm of the “chemically plausible”.
However, BuildAMol does know about some functional groups (which may be expanded upon in the future). Their purpose is to automatically generate a Linkage when connecting two molecules, without the user having to identify precicely which atoms they want to connect.
The catch is that BuildAMol only supports a handful of functional groups like this and due to its atom-substitution philosophy can only model substitution reactions but not addition reactions. That is to say, the limit is that the functional group needs to be able to loose some of it’s atoms in the reaction as leaving group (e.g. the OH that is lost as water from a carboxyl group when targeted by a nucleophilic attach).
Example#
Nothing beats a practical example to understand how we can use functional groups in BuildAMol to connect molecules. Let’s say we want to connect benzaldehyde to m-Toluidine like so:
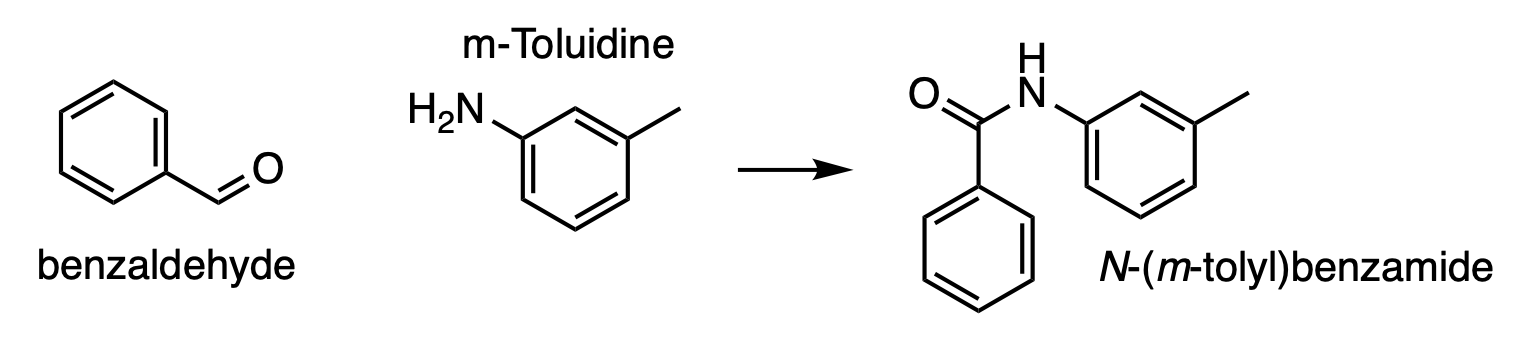
So we want to connect the aldehyde of benzaldehyde to the amine of m-Toluidine. Specifying the linkage manually would not be a big deal, especially if we autolabel them first. However, since both molecules only have one single aldehyde and amine respectively, we can quicken up the process by specifying that we want to connect the two molecules using the functional groups, and thus automatically infer the atoms involved in the linkage. Here’s how:
[1]:
import buildamol as bam
bam.visual.set_backend("py3dmol")
bam.load_small_molecules()
# first make the component molecules
benzaldehyde = bam.molecule("benzaldehyde")
toluidine = bam.molecule("m-toluidine")
Now that we have our component molecules, we can connect them. Instead of looking at them and identifying the atoms manually we will now use the functional groups to automatically generate the linkage. There is two ways we can do this:
Making a linkage with functional groups#
We can use the classmethod Linkage.from_functional_groups to generate a linkage which we can then use to connect the molecules. To do so, we need to specify which molecules we want to work with, and which functional groups to use. Functional groups can be found in the buildamol.structural.groups module.
[2]:
# we need to specify an electrophile and a nucleophile
# for this to work. emol / egroup are the molecule and functional group
# that serve as electrophile, and nmol / ngroup are the corresponding nucleophile.
link = bam.Linkage.from_functional_groups(
emol=benzaldehyde, egroup=bam.structural.groups.aldehyde,
nmol=toluidine, ngroup=bam.structural.groups.amine
)
print(link, link.bond, link.deletes, sep="\n")
Linkage(carbonyl_amine)
AbstractBond(atom1="C1'", atom2='N1', K=None, length=None)
([], [])
So, our linkage identified atoms C1 and N1 to be the atoms that will form a new bond. No atoms to delete were found since both atoms have hydrogen neighbors that can be removed as leaving groups.
[3]:
# now we can connect the components using our linkage
mol = benzaldehyde % link + toluidine
mol.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
And there we have our molecule.
But wait! What if I know which atoms to use on one molecule and only need to use one functional group to find the ones in the other molecule? Well, in this case we cannot use the from_functional_groups method to make the Linkage. Instead, we can use the functional group’s method infer_nucleophile_atoms or infer_electrophile_atoms directly to get the atoms we need from one molecule to then manually assemble the linkage we need. Here’s how:
[4]:
# let's say we want to connect the tuluidine via the methyl substituent
# and we know that the carbon is C7 but we need to find the right atoms
# for the aldehyde
# infer the atoms we need if we want the aldehyde to act as an electrophile
aldehyde_bonder, aldehyde_deletes = bam.structural.groups.aldehyde.infer_electrophile_atoms(benzaldehyde)
# now make the linkage we want
link2 = bam.linkage(aldehyde_bonder, "C7", aldehyde_deletes, [toluidine.get_hydrogen("C7")])
mol = benzaldehyde % link2 + toluidine
mol.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
And there we have another molecule!
Directly connecting with functional groups#
First making a linkage has the advantage of being able to verify what connection will be made before actually connecting the molecules. However, especially in cases like this where we can be fairly certain that no ambiguity or confusion is present with regard to which bonds will be made, we can use the method Molecule.react_with or the standalone function react to perform both linkage inference and molecule connecting in one go. Here’s how:
[5]:
# directly connect the two fragments without first making the linkage
mol2 = bam.react(
benzaldehyde,
toluidine,
egroup=bam.structural.groups.aldehyde,
ngroup=bam.structural.groups.amine
)
mol2.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
There we are again, same result! In fact, the molecular arithmetic syntax using operators also supports using functional groups. In this case we can use the % operator that is normally used to specify the linkage to specify the functional groups to use. However, in this case we need to make sure to specify it for both molecules involved! Here’s the same example as above using arithmetic syntax:
[6]:
# specify the functional groups directly using % on BOTH molecules
# in this case the first molecule is always the electrophile and the second is the nucleophile
mol2 = benzaldehyde % bam.structural.groups.aldehyde + toluidine % bam.structural.groups.amine
mol2.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
A word of warning. This type of pseudo-reaction works on residue level. It requires that the attach_residue of the participating molecules only have one functional group of the kind that is used to make the connection. If multiple ones are present, the ambiguity may lead to unpredictable behaviour when connecting molecules!
Custom Functional Groups#
So far we have looked at how we can use pre-existing functional groups to connect molecules. But what if we need a functional group that does not ship with BuildAMol? We can define our own functional group in these cases.
Let’s say we want to make our own nitro functional group. Here’s how we can do it:
[7]:
# we start by making a new functional group object
# and provide data on the atom connectivity that describe the nitro group
nitro = bam.structural.groups.FunctionalGroup(
id="nitro",
# rank is the "importance" of the group, higher ranks will replace lower ranks when multiple groups would match a certain set of atoms
rank=2,
# the geometry that describes the spacial arrangement of atoms
# around the central atom. The nitro group is planar around
# the N=O center
geometry=bam.structural.geometry.trigonal_planar,
# the participating atoms that define what a nitro group is are
# (the first atom is the geometric center, i.e. the N)
# (only direct neighbors of the central atom are included here!)
atoms=("N", "O", "O"),
# the bonds that connect the atoms in the atoms list above
# these numbers define the bond orders of the atoms to the center atom
# (e.g. the bond of N to the first O is a double bond (order 2) and to
# the second O is a single bond). This is a list with 'atoms'-1 entries
# (because we don't have a bond from the central atom to itself)
connectivity=(2, 1),
# now we may need to provide some additional constraints on the atoms'
# chemical neighborhood (i.e. extended connectivity)
# this is a list with one entry for each atom in 'atoms'.
# entries can be None (no constraints, rare), or any function
# from the 'constraints' module that returns a boolean value
constraints=[
# the central N needs to have exactly 3 neighbors
bam.structural.neighbors.constraints.has_n_neighbors(3),
# the first O needs to be only connected to N (i.e. have 1 neighbor)
bam.structural.neighbors.constraints.has_n_neighbors(1),
# finally, the second O must have two neighbors which are exactly the
# central N and a hydrogen (we need the hydrogen to use this O as a bonder, later on...)
bam.structural.neighbors.constraints.neighbors_exactly("N", "H")
]
)
Well, that’s a pretty large cell, but luckily most of it is comments. We have now successfully set up our nitro group. However, we cannot yet use it to make linkages. This needs to be done in the next step where we specify which atoms to use for making bonds and which ones to delete. This is done using the set_reactivity method which allows us to specify how the group will behave if it is used as an electrophile or a nucleophile. We can specify both behaviours or only one, depending on our
needs.
Just as a reminder again, since BuildAMol does not follow chemical conventions, there is no need to specify chemically plausible behaviour, it is simply about which atoms to use for making bonds and which atoms to remove, that’s all.
Here is the reactivity that we want to achieve:
Electrophile#
As an electrophile, the incoming molecule should connect to the central N and the nitro group should loose an OH in the process to achieve an R1-(N=O)-R2 link.
Nucleophile#
As a nucleophile, we want the nitrogroup’s second O to bind to the incoming molecule, whereby it losses simply it’s hydrogen neighbor in the process, to result in R1-(N=O)-O-R2.
[8]:
nitro.set_reactivity(
# the index of the atom (in the 'atoms' list from the above definition)
# that should bind in case of the group being the electrophile (i.e. the central N)
electrophile_bonder=0,
# the second O should bind in case of being the nucleophile (i.e. the one with the single bond N-O)
nucleophile_bonder=2,
# now we define what atoms to loose. We can either specify a list of indices like above or we can
# specify a function that will generate a list of atoms.
# We will do the latter and define the following function to get
# the second O atom (index 2) as well as it's hydrogen neighbor...
electrophile_deletes=lambda mol, res, bonder, atoms: [
atoms[2], mol.get_hydrogen(atoms[2])
],
# no need to specify anything for the nucleophile deletes
# because loosing a hydrogen neighbor is the default behaviour
# anyway...
)
[8]:
FunctionalGroup(nitro)
Now we are finally done with all the setup… We can now use our nitro group to make linkages and connect molecules. Let’s try it out…
[9]:
# make a molecule with a nitro compound
nitro_benzene = bam.read_smiles("C1=CC=C(C=C1)[N+](=O)[OH]")
nitro_benzene.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
Now we can connect this nitro benzene to our benzaldehyde using only the functional groups. Like so:
[10]:
mol2 = bam.react(
benzaldehyde, nitro_benzene,
egroup=bam.structural.groups.aldehyde,
ngroup=nitro # our nitro group acts as the nucleophile
)
mol2.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
Or alternatively, to use our nitro group as electrophile we could do something like:
[11]:
mol3 = bam.react(
nitro_benzene, toluidine,
egroup=nitro, # our nitro group is the electrophile
ngroup=bam.structural.groups.amine
)
mol3.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
And like that we made our own custom functional group that we can use to connect molecules together.
With that we have reached the end of this tutorial. We saw how we can use functional groups to connect molecules together without needing to manually specify the atoms involved in the linking. We also so went over making custom functional groups to tailor to our own needs.
Thank’s for checking out this tutorial and good luck with your research using BuildAMol!