Creating Pseudo-Reactions#
In this tutorial we will cover:
how we can use the
Reactionclass to define linkages without identifying the participating atoms directly
BuildAMol lives on the idea of connecting two molecules by establishing a bond between two atoms and removing (at least) to others in exchange. This allows us to create aribtrary structures irrespective of chemical constraints.
Creating a Linkage is usually not difficult but requires some manual intervention, e.g. looking at the topology and identifying the right atom ids and residues. Functionalities such as functional groups, constrained atom searching, or automated downstream atom deletion provide some support to at least partially automate the process of identifying the right atoms to use when defining a linkage. However, they usually are either relatively constrained in their applicability (e.g. functional
groups) or still require some additional boilerplate code to ultimately assemble finished molecules.
The Reaction class provides a highlevel hub for exactly such boilerplate code to define linkages and connect molecules together. The input for the class are similar to that of a Linkage except that instead of providing individual atoms we can provide function that will be applied to dynamically obtain the right atoms from molecules when applying the reaction. Therefore Reactions are more powerful versions of the classic Linkage.
The
Reactionclass is part of the core BuildAMol library as of version1.2.11. We can therefore directly use it asbam.Reactionafter importing.
Let’s check out how we can use it!
Example 1 - Benzene + EtOH#
To start simple let’s have ethanol perform an addition on a benzene ring. Our reaction mechanism should have the Oxygen perform attack an aromatic Carbon (with standard hydrogen-deletion). Remember, this is still BuildAMol so chemical reasoning is still not relevant when defining a “Reaction”!
[1]:
from warnings import filterwarnings
filterwarnings("ignore", category=DeprecationWarning)
import buildamol as bam
bam.load_small_molecules()
# start with getting the compounds
bnz = bam.get_compound("benzene")
etoh = bam.get_compound("ethanol")[0]
/opt/anaconda3/lib/python3.12/site-packages/jupyter_client/session.py:203: DeprecationWarning: datetime.datetime.utcnow() is deprecated and scheduled for removal in a future version. Use timezone-aware objects to represent datetimes in UTC: datetime.datetime.now(datetime.UTC).
return datetime.utcnow().replace(tzinfo=utc)
Now we can define the Reaction we want to perform. To do so we need to define (at least) two functions:
A function to obtain
atom1(i.e. the target atom) that takes aMoleculeas only argumentA function to obtain
atom2(i.e. the source atom) that takes aMoleculeas only argument(optional) A function to obtain the atoms to
delete_in_targetthat takes at least aAtomas argument (will receiveatom1) and can optionally receive theMoleculeas second argument.(optional) A function to obtain the atoms to
delete_in_sourcethat follows the same rules…
Once we have at least 1 and 2, we can set up our reaction. For our purposes the reaction could look like this:
[2]:
reaction = bam.Reaction(
atom1 = lambda mol: mol.get_atoms("C", by="element").pop(), # get any carbon (since all are aromatic in benzene)
atom2 = lambda mol: mol.get_atoms("O", by="element").pop(), # get any oxygen (since ethanol only has one)
)
We don’t need to define deletion atoms since Hydrogen-deletion is fine for now. To apply the reaction we can either call the reaction with molecules or use the apply method.
[3]:
out = reaction(bnz, etoh)
# or
# out = reaction.apply(bnz, etoh)
out.draw2d().draw(height=200, width=200)
/opt/anaconda3/lib/python3.12/site-packages/buildamol/utils/visual.py:164: DeprecationWarning: The `highlight_color` argument is deprecated and will be removed in future versions. Please specify a color when calling `highlight_atoms` or `highlight_bonds` instead.
aux.deprecation_warning(
/opt/anaconda3/lib/python3.12/site-packages/buildamol/utils/visual.py:168: DeprecationWarning: The `linewidth` argument is deprecated and will be removed in future versions. Please specify a linewidth when calling the `draw` method instead.
aux.deprecation_warning(
[3]:
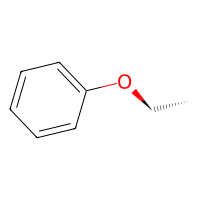
We can now actually reuse the same reaction to connect other molecules as well, as long as the atom functions are applicable to them.
[4]:
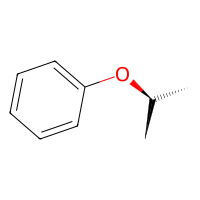
# get another alcohol
isopropanol = bam.molecule("isopropanol")
out2 = reaction(bnz, isopropanol)
out2.draw2d().draw(height=200, width=200)
[4]:
Example 2 - Extended Leaving Group#
To make things a little more complex let’s try making two functions now, one for adding and one for removing a tosylate group.
[5]:
tso = bam.molecule("Tosylate").add_hydrogens() # to make the SO3 group have an OH instead of O-
tso.rename_residue(1, "TSO")
phenol = bam.hydroxylate(bnz, 1) # first make phenol from benzene
[6]:
from buildamol.structural import constraints_v2 as constraints
add_tosylate = bam.Reaction(
# in the target molecule we want to connect to some hydroxyl oxygen
atom1 = lambda mol: mol.get_atoms("O", by="element", filter=constraints.has_neighbor_hist({"H": 1, "C": 1})).pop(),
# in the tosylate (source) we want to connect to the Sulfur
atom2 = lambda mol: mol.get_atoms("S", by="element").pop(),
# we then want to get rid of the OH from the TsO
delete_in_source = lambda atom: atom.get_neighbors(filter=lambda a: a.element == "O" and len(a.get_hydrogens()) > 0).pop(),
)
# now we can use this to add tosylate groups
out3 = add_tosylate(phenol, tso)
out3.draw2d().highlight_residues(1, color="yellow").draw(height=200, width=200)
[6]:
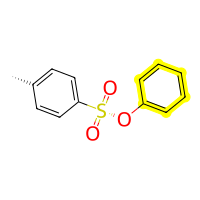
And now that we have a tosylated phenole we can define a function to remove the tosylate again when having another reactant perform a nucleophilic attack on the C-O-TsO. This will involve a little more code for the functions we need to define but bear with and hopefully it will be clear how the setup works!
[7]:
# in the target molecule we want to connect to some carbon
# with an oxygen neighbor that neighbors a tosylate group
def get_attackable_carbons(mol):
tso_sulfurs = mol.get_atoms("S", by="element", filter=lambda a: a.parent.name == "TSO")
carbons_at_distance_2 = set()
for sulfur in tso_sulfurs:
neighbors = sulfur.get_neighbors(n=2, mode="at", filter=lambda a: a.element == "C" and a.parent.name != "TSO")
carbons_at_distance_2.update(neighbors)
return list(carbons_at_distance_2)
# for each attacked carbon we want to remove the tosylate group
# with automatic downstream deletion it is enough to identify the right oxygen
def get_tosylate_atoms(carbon):
oxygen = carbon.get_neighbors(
filter=lambda a: a.element == "O" and any(n.parent.name == "TSO" for n in a.get_neighbors())
)
if len(oxygen) == 0:
raise ValueError("No tosylate oxygen found")
return oxygen.pop()
# now we still need some mechanism for the attacking molecule
# to keep things simple on this side let's simply use an amine that performs a nucleophilic attack
def get_amine_nitrogen(mol):
return mol.get_atoms("N", by="element", filter=constraints.has_neighbor_hist({"C": 1, "H": 2})).pop()
# with all of this we can now define the reaction
remove_tosylate = bam.Reaction(
atom1 = get_attackable_carbons,
atom2 = get_amine_nitrogen,
delete_in_target = get_tosylate_atoms,
)
[8]:
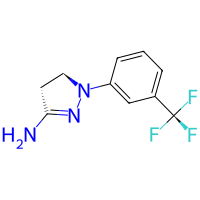
# let's get some primary amine from pubchem
amine = bam.molecule("BW-755C")
amine.draw2d().draw(height=200, width=200)
[8]:
[9]:
# and let it replace the tosylate group
out4 = remove_tosylate(out3, amine)
out4.draw2d().highlight_residues(1, color="yellow").draw(height=200, width=200)
[9]:
This may look like a lot more effort than simply going and defining Linkages directly and if we are only going to modify molecules on a case-to-case basis then defining entire reactions is probably not worth the time. However, if your workflow involves repeadetly performing the same operations you will likely appreciate the power of the Reaction class.
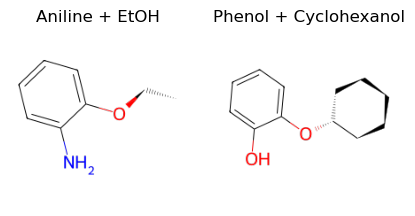
Just to prove the concept that our function also works on other molecules let’s also play with a second set of molecules:
[10]:
A = bam.molecule("aniline")
B = bam.molecule("cyclohexanol")
outA = reaction(A, etoh)
outB = reaction(phenol, B)
import matplotlib.pyplot as plt
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(5, 4))
ax1.imshow(outA.draw2d().draw(ax=ax1, height=200, width=200))
ax2.imshow(outB.draw2d().draw(ax=ax2, height=200, width=200))
ax1.set_title("Aniline + EtOH")
ax2.set_title("Phenol + Cyclohexanol")
ax1.axis("off")
ax2.axis("off")
/opt/anaconda3/lib/python3.12/site-packages/buildamol/utils/visual.py:164: DeprecationWarning: The `highlight_color` argument is deprecated and will be removed in future versions. Please specify a color when calling `highlight_atoms` or `highlight_bonds` instead.
aux.deprecation_warning(
/opt/anaconda3/lib/python3.12/site-packages/buildamol/utils/visual.py:168: DeprecationWarning: The `linewidth` argument is deprecated and will be removed in future versions. Please specify a linewidth when calling the `draw` method instead.
aux.deprecation_warning(
/opt/anaconda3/lib/python3.12/site-packages/jupyter_client/session.py:203: DeprecationWarning: datetime.datetime.utcnow() is deprecated and scheduled for removal in a future version. Use timezone-aware objects to represent datetimes in UTC: datetime.datetime.now(datetime.UTC).
return datetime.utcnow().replace(tzinfo=utc)
[10]:
(-0.5, 199.5, 199.5, -0.5)
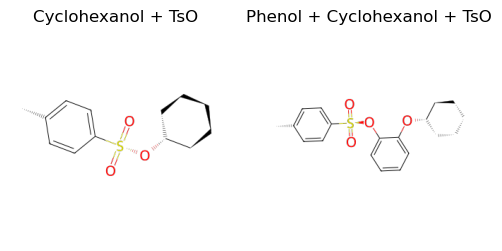
Let us also tosylate the cyclohexanol and outB.
[11]:
tso_B = add_tosylate(B, tso)
tso_outB = add_tosylate(outB, tso)
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(6, 9))
v_B = tso_B.draw2d().draw(height=300, width=300)
v_outB = tso_outB.draw2d().draw(height=300, width=300)
ax1.imshow(v_B)
ax2.imshow(v_outB)
ax1.set_title("Cyclohexanol + TsO")
ax2.set_title("Phenol + Cyclohexanol + TsO")
ax1.axis("off")
ax2.axis("off")
[11]:
(-0.5, 299.5, 299.5, -0.5)
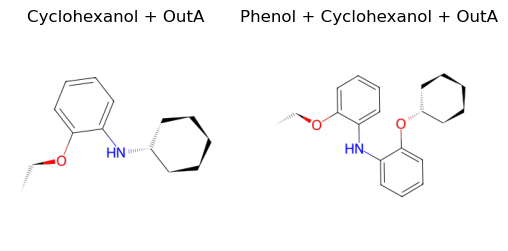
And finally use outA to remove the tosylate group again.
[12]:
final_B = remove_tosylate(tso_B, outA)
final_outB = remove_tosylate(tso_outB, outA)
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(6, 9))
v_B = final_B.draw2d().draw(height=300, width=300)
v_outB = final_outB.draw2d().draw(height=300, width=300)
ax1.imshow(v_B)
ax2.imshow(v_outB)
ax1.set_title("Cyclohexanol + OutA")
ax2.set_title("Phenol + Cyclohexanol + OutA")
ax1.axis("off")
ax2.axis("off")
[12]:
(-0.5, 299.5, 299.5, -0.5)
Multiple target sites#
So far we have had reactions perform a single operation each time. However, we can also have a reaction perform multiple additions at multiple target sites simultaneously. To do so, we simply have to let the atom1 function return an iterable of multiple Atom objects (e.g. a tuple, list, or set).
[15]:
# actually, our add tosylate function already checks for all possible hydroxyl groups
# but then simply pops the first one it finds
# to change this we can either redefine the Reaction using the `set` method or we create a copy
# of it with a modified atom1 function using the `with_reactivity` method.
# let's get a new reaction that checks all hydroxyl groups
multiple_site_add_tosylate = add_tosylate.with_reactivity(
# this time we don't pop at the end so we get all possible hydroxyl oxygens
atom1 = lambda mol: mol.get_atoms("O", by="element", filter=constraints.has_neighbor_hist({"H": 1, "C": 1})),
)
C = bam.read_smiles("OCCCO")
# and add tosylate groups
outC = multiple_site_add_tosylate(C, tso)
outC.draw2d().highlight_residues(1, color="yellow").draw(height=200, width=500)
[15]:
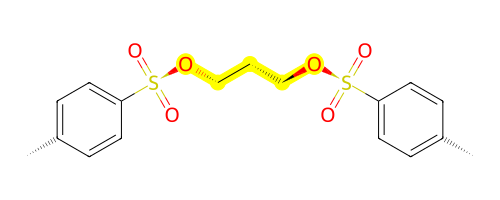
And there we have it, two tosylate groups added together!
And with that we have reached the end of this tutorial on the Reaction class! As we have seen we can define reusable and flexible reaction mechanisms by simply defining functions that find the right atoms to participate in defining a linkage.
Thanks and good luck with your research project using BuildAMol!