Getting Started#
Welcome to the first BuildAMol tutorial, where we shall learn the basics of operating with BuildAMol!
In this tutorial we will cover:
which (main) classes and modules exist and when to use them
how to read input to make
Moleculeshow to connect two molecules together
Main Classes in BuildAMol#
The most important class is the Molecule class which houses ~90% of all functionality that the average user is likely to use. Next, comes the Linkage class that defines how multiple molecules can be connected, so the user can build larger structures. Third are the ptimization environments of the optimizers module. They can be used to optimize conformations. Also, the MoleculeViewer3D may come in quite handy. It is essentially just a plotly 3D-plot but it can be selectively
colored easily so as to highlight specific parts of your molecule - it is a great tool to check if your building process is working (or to debug why it may not be). Finally, the resources module houses a wealth of reference structures for users to use when building their favourite structures.
That’s already about it! Of course, there is a lot more - but everything else is mostly underneath the surface and will most likely not bother the average user directly.
[16]:
import plotly
plotly.offline.init_notebook_mode()
import buildamol as bam
Resources#
BuildAMol includes the PDBE component library for small molecules (or at least a pretty large part of it). Since this is quite large, BuildAMol will not by default load any data, but offers a buch of functions for the user to load what they are interested in using selectively. We can also set defaults that are always loaded if we want to re-use our own custom builds in different sessions.
Available pre-loadable sets are:
# to load the amino acid and derivative compounds
bam.load_amino_acids()
# to load nucleotide and derivative compounds
bam.load_nucleic_acids()
# to load sugar and derivative compounds
bam.load_sugars()
# to load miscallaneous small molecules of less than 40 atoms
bam.load_small_molecules()
Molecules#
Molecules are the essential data unit in BuildAMol. Each Molecule houses atoms that form a molecular structure. We can generate Molecules from:
a PDB or CIF file (e.g. “my_structure.pdb”)
a PDB ID (e.g. “GLC” - the PDB id for alpha-D-glucose)
a trivial name (e.g. “triacetamide”)
a chemical formula (e.g. “C2H6O” - caution, may produce ambiguities!)
SMILES and InChI/InChIKey
… and more
The Molecule class has classmethods for all of these named from_pdb, from_compound (for PDB ID, name etc.), from_pubchem and so forth. But there also exists the toplevel molecule function that will try to automatically figure out the user input and generate a molecule for you.
[17]:
# make sure we have amino acids available
bam.load_amino_acids()
# get a serine
ser = bam.molecule("serine")
# check what it looks like
ser.show3d()
/opt/anaconda3/lib/python3.12/site-packages/jupyter_client/session.py:203: DeprecationWarning:
datetime.datetime.utcnow() is deprecated and scheduled for removal in a future version. Use timezone-aware objects to represent datetimes in UTC: datetime.datetime.now(datetime.UTC).
We can always check out if a compound is available in a loaded set using the has_compound function. But even if a specific compound is not available BuildAMol can retrieve the molecular structure directly from PubChem to guarantee a smooth workflow!
[18]:
# check if we have diacetamide
bam.has_compound("diacetamide")
[18]:
False
[19]:
# get a diacetamide (using its name)
# since diacetamide was not available in the loaded set of compounds
# BuildAMol will automatically download it from PubChem
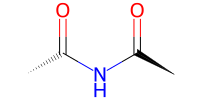
tri = bam.molecule("diacetamide")
# check what it looks like
# this time with a 2D depiction (use 'draw' to set some parameters,
# use 'show' otherwise as a shortcut)
tri.draw2d().draw(width=200, height=100)
[19]:
Linkage#
Now assuming we want to take diacetamide and connect the serine to it. We can define a Linkage to specify which atoms to link between the two. To do so, we need to know what the atoms are labelled as (another reason to always visualize the structures). In the Linkage definition we specify, for instance, that the diacetamide-N1 should be connected to serine-C. In the process, we remove the a hydrogen from the acetamide’s N1 and the serine’s OXT and HXT.
Note
The linkages do not need to follow chemical reasoning - they only connect structures by adding and removing connections. Hence, we could connect the molecules by one of the methyl-groups making one of the hydrogens the leaving group (even though chemically such a reaction would probably never occur) - BuildAMol builds structures, but it does not imitate chemical reactions!
[20]:
# define the linkage between the two molecules
# remember to check the atom labelling as these atom labels must match
# the existing atom labels in both molecules
link = bam.linkage(atom1="N1", atom2="C", delete_in_target=["H1"], delete_in_source=["OXT", "HXT"])
Now we can connect the two molecules for example using the connect function:
[21]:
# connect the two molecules
new = bam.connect(tri, ser, link)
# and check what it looks like
# we can also use different backends, e.g. py3dmol
new.py3dmol().show()
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
BuildAMol also supports operator-based syntax:
[ ]:
# equivalent to the above, % specifies the linkage, + connects the two molecules
new = tri % link + ser
new.py3dmol().show()
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
Pseudo-Reactions#
Even though chemical rationale is by no means part of BuildAMol’s core idea, to facilitate the building process and offer some aid for users who strong think in chemical terms, BuildAMol offers a mechanism to imitate reactions to some degree by providing Reactivity patterns for functional groups and a Reaction class to automatically detect matching atoms to use for linking and removal.
To repeat the above example, we had an imide react with a carboxyl group. The imide itself is not actually a defined reactivity (yet?) but since it consists basically of 1.5 amide groups we can borrow the reactivity of the amide group for our purposes.
[22]:
from buildamol.structural.reactivity import Amide, Carboxyl
# define the reaction
reaction = bam.Reaction.from_reactivities(nucleophile=Amide(), electrophile=Carboxyl())
# connect the two molecules using the reaction
new2 = reaction(ser, tri)
new2.draw2d()\
.highlight_bonds(new2.get_residue_connections(), color="orange")\
.draw(width=300, height=200)
[22]:
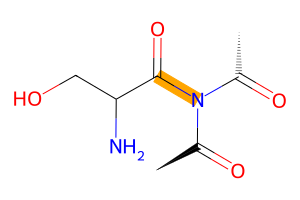
By the way, these kinds of visualizations (especially the 2D one) can be easily customized in various ways (check out the tutorial for that to learn more).
Optimization#
If we observe a clash in the resulting conformation, we can apply an optimization. This is where the optimizers module comes into play. BuildAMol uses a rotational optimization system to find conformations without the need for molecular dynamics. This can be efficient or very expensive (up to unsolvable, in fact!) depending on the pre-processing / pre-thinking that the user provided. But don’t worry, just don’t try to optimize hundreds of bonds at once (that won’t work, trust me, I’ve
tried!).
The short way of optimizing a molecule is by using it’s optimize method. This method does not require any additional arguments, although we can pass further instructions. If you are interested, go check out the tutorial on optimization to learn more!
[23]:
# prepare a 3D drawing with plotly (default backend) before optimization
v = new.plotly(atoms=False)
# optimize the new molecule
new = new.optimize()
v.add(new.plotly(line_color="red", atoms=False))
v.show()
/opt/anaconda3/lib/python3.12/site-packages/jupyter_client/session.py:203: DeprecationWarning:
datetime.datetime.utcnow() is deprecated and scheduled for removal in a future version. Use timezone-aware objects to represent datetimes in UTC: datetime.datetime.now(datetime.UTC).
Formats and Integrations#
Now that we have a structure we like we can for instance save it as a PDB or mmCIF file. If we have more intricate interests in the molecules we have built we can export them directly to rdkit or openbabel objects for further analysis.
To make working with different data formats as easy as possible, each BuildAMol Molecule can be created and saved to the following formats. Here are is a table with the corresponding methods available in the Molecule class:
Format |
As input |
As output |
|---|---|---|
PDB |
|
|
mmCIF |
|
|
Molfile / SDF |
|
|
SMILES |
|
|
JSON |
|
|
XML |
|
|
XYZ |
|
|
PDBQT |
|
|
pickle |
|
|
rdkit |
|
|
openbabel |
|
|
openmm |
|
|
stk |
|
|
biopython |
|
|
[24]:
# so let's save our small molecule to a PDB file
new.to_pdb("./files/our_first_molecule.pdb")
Beyond connecting Molecules#
We can also perform a plethora of other operations on molecules beyond stitching them together in creative ways. BuildAMol can modify existing molecules quickly in almost any way imaginable.
Need to replace one functional group with another? No problem!
Want to change the element of one atom? Sure!
Turn a cis double bond to trans? Of course!
Align a molecule in space relative to something else? Yep!
With that we have reached already the end of this first little introduction into what BuildAMol can do, and how it feels to work with BuildAMol. Thanks a lot for reading this far! And good luck with whatever you may do with BuildAMol in your projects!